Publication
693
Inorg. Chem. 50 (20), 10190-10203, 2011
DOI: 10.1021/ic201168j
|
|
|
|
|
 |
Geometric and Electronic Structures of Peroxomanganese(III) Complexes Supported by Pentadentate Amino-Pyridine and -Imidazole Ligands |
|
|
|
|
Robert A. Geiger, Domenick F. Leto, Swarup Chattopadhyay, Pierre Dorlet, Elodie Anxolabéhère-Mallart, and Timothy A. Jackson
Department of Chemistry and Center for Environmentally Beneficial Catalysis, University of Kansas, Lawrence, Kansas 66045, United States
Laboratoire Stress Oxydant et Detoxication, CNRS URA 2096 and CEA/iBiTec-S/SB2SM, CEA Saclay, 91191 Gif-sur-Yvette Cedex, France
Laboratoire d’Electrochimie Moléculaire, UMR 7591 CNRS, Université Paris Diderot, Sorbonne Paris Cité, 15 rue Jean-Antoine de Baïf, F-75205 Paris Cedex 13, France
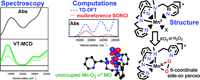
Three peroxomanganese(III) complexes [MnIII(O2)(mL52)]+, [MnIII(O2)(imL52)]+, and [MnIII(O2)(N4py)]+ supported by pentadentate ligands (mL52 = N-methyl-N,N',N'-tris(2-pyridylmethyl)ethane-1,2-diamine, imL52 = N-methyl-N,N',N'-tris((1-methyl-4-imidazolyl)methyl)ethane-1,2-diamine, and N4py = N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine) were generated by treating Mn(II) precursors with H2O2 or KO2. Electronic absorption, magnetic circular dichroism (MCD), and variable-temperature, variable-field MCD data demonstrate that these complexes have very similar electronic transition energies and ground-state zero-field splitting parameters, indicative of nearly identical coordination geometries. Because of uncertainty in peroxo (side-on ?2 versus end-on ?1) and ligand (pentadentate versus tetradentate) binding modes, density functional theory (DFT) computations were used to distinguish between three possible structures: pentadentate ligand binding with (i) a side-on peroxo and (ii) an end-on peroxo, and (iii) tetradentate ligand binding with a side-on peroxo. Regardless of the supporting ligand, isomers with a side-on peroxo and the supporting ligand bound in a tetradentate fashion were identified as most stable by >20 kcal/mol. Spectroscopic parameters computed by time-dependent (TD) DFT and multireference SORCI methods provided validation of these isomers on the basis of experimental data. Hexacoordination is thus strongly preferred for peroxomanganese(III) adducts, and dissociation of a pyridine (mL52 and N4py) or imidazole (imL52) arm is thermodynamically favored. In contrast, DFT computations for models of [FeIII(O2)(mL52)]+ demonstrate that pyridine dissociation is not favorable; instead a seven-coordinate ferric center is preferred. These different results are attributed to the electronic configurations of the metal centers (high spin d5 and d4 for FeIII and MnIII, respectively), which results in population of a metal-peroxo s-antibonding molecular orbital and, consequently, longer M–Operoxo bonds for peroxoiron(III) species. |


